|
第二个是“读懂”,即找出全部基因在染色体上的位置,了解它们的功能。
整个基因组测序完成后的数据可以构成一本100万页的书,其上只有4个字母的反复出现.
如何处理、存储和分析这些数据?这已不是生物学家本身可以解决的问题,需要其他学科,特别是数学与计算机学科的介入.。首先介绍了分子生物学的一些最基本的知识,然后着重介绍了目前人类基因组研究中的若干问题及其所用到的数学方法与模型。
1、背景与基本知识
生命的基本单位是细胞,它由细胞膜、细胞质和细胞核三者组成,遗传信息储存在细胞核中。人的细胞核中含有23对染色体,染色体含dna(脱氧核糖核酸)和蛋白质。dna经螺旋、扭曲、折叠等压缩到万分之一并与蛋白质一起而组成染色体。
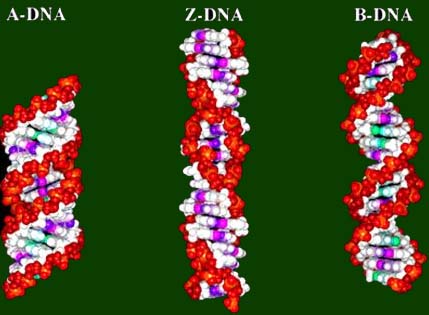
dna是一种大分子,由两股长链以螺旋式构成,这种螺旋结构是在1953年由沃森和克里克提出,并获1962年的nobel奖,是20世纪最伟大的科学发现之一。
dna分子上的一个个有生物功能的片段是基因。基因由若干按一定顺序排列的核苷酸组成。核苷酸由磷酸基团、脱氧核糖及碱基构成,有4种不同的碱基,即:腺嘌呤、鸟嘌呤、胞嘧啶及胸腺嘧啶,分别用a,
g,c,t表示。核苷酸按其所含碱基的不同也分为4种。在dna的双股上,a,t成对出现,g,c也成对出现,每对称为一个碱基对。
遗传密码在dna的链上,密码由4种不同的核苷酸按一定顺序排列而成,即可看成由4个字母a,g,c,t排列而成.
据估计,人类的dna约含有30亿个密码,排列组成至少10万条基因.
决定人体蛋白质的20种氨基酸的遗传密码已找出,先由a, g, c,
t中每3个字母重复排列成一密码子,共有43=64个密码子.
每一密码子对应一种氨基酸;但由于只有20种氨基酸,故这种对应只能是多对一的,例如aga,agg都对应于精氨酸,此种对应关系已完全确定,称为遗传密码字典。
使人们惊叹不已的是,自然界所有的生命形式都共用这本密码。在确定了三联体码在dna上线性串接的结合方式后,发现了为蛋白质编码的基因结构。这些基因在dna上所处的位置,称为dna的编码区,约占整个基因组的3%—5%,其余部分习惯上统称为“废物(junk)”dna.
在对编码区上dna的结构所进行的40多年的研究工作中,已造就了几十名nobel奖获得者。然而,“junk”dna中包含的信息也许更多。总之,细胞细胞核染色体蛋白质(含氨基酸)、dna(由核苷酸组成)基因(上有密码子,即由a,g,c,t组成的三联体码)。
1987年,美国开始了人类基因组研究计划,任务有两个:第一个是“读出”,即研究出人类基因组的全部核苷酸的顺序;第二个是“读懂”,即找出全部基因在染色体上的位置,了解它们的功能。用数学的语言来说,人类基因组计划的最基本、最直接的结果是得到一个由4个字母(a,g,c,t)可重复排列而组成的长度为3×109的一维链.
解读后,人们不仅获得静态的结构信息,而且还能得到动态的四维(时空)调控信息.
目前国际上找到了全长基因约2万条, 平均每天能找到9条.
据报道,复旦大学发明了一种新技术,每日能找到15条.
科学家们把此计划与40年代的曼哈顿计划(研究原子弹、氢弹)和60年代的阿波罗计划(宇航、登月)相比。
2、目前基因组研究中的若干数学方法
(1 )概率统计方法
概率统计是较早进入生命科学研究领域的学科之一,早在20世纪40—50年代,fisher和wright就用它研究过数量遗传学。下面给出几个例子说明它在当今人类基因研究中的应用。
※2.1.1 隐马尔可夫模型(hmm)
隐马尔可夫模型(hidden markov
models)是由两个随机变量序列组成:一个是观测不到的马尔可夫链{yn: n≥0},
另一个是可以观测到的随机序列{xn: n≥0}。且已知两者间有如下的联系:
n,条件概率为已知。{yn, n≥0}称为隐马尔可夫链,{xn, n≥0}称为其观测链。
隐马尔可夫模型已在语音识别中得到广泛应用[2,3],80年代末开始应用于计算生物学。目前,隐马尔可夫模型在人类基因组研究的许多方面都有广泛的应用,如,dna序列的阵排列(alignment)[4~6]、寻找基因(genefinding)[7,8]、作基因图(genetic
mapping)[9]、作物理图(physical
mapping)[10]及蛋白质二级结构的预测[11]等.
这诸多应用的基本思想都源于如下的bayes统计分析:
考虑随机模型m=(s,pm), 对分别以p(m | s), p(s | m)表示p(x来自模型m
| x=s), p(x=s |
x来自模型m);则上述诸应用问题大多可抽象为如下的识别问题或判别问题——
(1)识别问题:现观察到一序列s,
问此序列是来自模型m的概率是多少?由bayes公式,欲计算p(m |
s),需知道两个先验概率p(m)和p(s)。但在实际问题中往往很难做到.
可退一步考虑如下的判别问题。
(2)判别问题:再考虑另一随机模型n=(s,pn),根据观察到的序列s,
判断s是来自m,还是来自n?此时,可由比较两个条件概率p(m | s)和p(n |
s)的大小来得到答案。仍由bayes公式可得。
下面以“多个dna序列的阵排列问题”为例子说明如何在dna序列上建立隐马尔可夫模型(hmm)。
设有r个dna序列,其中, 序列a1, a2, …,ar的一个阵排列定义为:在a1, a2,
…, ar中不同的位置分别插入空隙符号“-”(gap),使其变为l≥max{n1, …,
nr}, 显然,对于给定的序列a1, a2,…,
ar可以有许多不同的阵排列。例如,都是序列a=aggt, b=atgtg的阵排列。
引入阵排列的目的在于讨论序列之间的相似性,为此,定义函数和要寻找使d(a1, a2, …,
ar)达到最小的那些排列,称其为“最优的阵排列”。由于所有可能的阵排列的数目随序列的长度及序列个数呈指数增加,例如当r
= 2, n1 = n2 = l = 1
000所有可能的阵排列的数目大约为10600[12]。直接进行逐个比较来得到最优的阵排列是不现实的,必须寻找其他的算法。最近,用hhm来解决此问题,得到了较好的结果(见文献[4,13])。隐马尔可夫模型(hmm)可如下建立:隐马尔可夫链yn取值为m(配对)、i
(插入)、及d(删除)3个状态;可观察到的序列xn取值为a,g,c,t.
隐马尔可夫链yn可以看做在dna序列上运动,在第k步时(相应于dna序列上的第k个位置)取值若为配对状态m,则以概率pk(a
| m)在此位置产生字母若为插入状态i,则以概率pk(a | i)在此位置插入字母
若为删除状态d,则将位置k上的字母删除。隐马尔可夫链yn的转移矩阵为p( yk 1 | yk
)。于是当隐马尔可夫链yn运动了n 1步后,便可得到两个序列,一是状态序列:y0(开始),
y1, …,yn, yn 1 = ml 1(结束)(观察不到);二是字母序列: x1, x2,
…, xv, v≤n(可观察到)。当yi是m(配对)或i(插入)时,产生的字母记为xli,
则状态序列y=y0,y1,…,yn,yn 1与字母序列x = x1, x2, …,
xv的概率分布为(其中, 因此序列x = x1, x2,…,
xv出现的概率为)。利用训练集可以将模型中的未知参数k=0,…, n估计出来。
※2.1.2 物理图与随机区间覆盖问题
作图是人类基因组研究计划中一项主要的任务,通过作图可确定基因及其他功能区在dna序列上的位置。关键的图是物理图和遗传图,遗传图是通过关联分析将已知的基因的相对位置定位在染色体上;物理图是将一组标记定位在染色体上并估计其间的相互距离,每一标记点可以是一条已知基因,也可以不是。现在,已构造出具有
30 000个标记点的整个人类基因组的物理图,每个标记点的平均距离大约是100 kb
(kilobases)。
在作物理图的过程中遇到如下的随机区间覆盖问题:
设m是一给定的区间(相当于染色体),其长度为g;i为随机区间之集,其元素的区间长度的分布已知;p是随机点的集合,其点随机地分布在区间m上。称一区间为anchored,如果它至少包含p中一个元素,称i中的两区间为连接的,如果它们的交集中包含p中一点,将所有相互连接的区间以它们所包含p中的最小点为左端点,以它们所包含p中的最大的点为右端点,组成一个新区间。称为一个重叠群
(contig)。问题是:应选取多少个i中的元及p中的元,才能使所有的重叠群几乎覆盖了区间m?譬如,覆盖m的比例平均起来达99%.
此问题已由arratia等[14]较为圆满的解决。
※2.1.3 结肠癌与大偏差医学上发现 结肠癌是一种遗传因素占主导地位的疾病,在某些家族中发病率很高,并有继承性.
1991年kinzler等人报告(见文献[13]),结肠癌与位于第5条染色体长臂上的,称做apc的遗传基因的变异有关。但后来进一步的研究表明,同样都是apc基因变异的人,而受感染的程度却大不相同。一年后,dove及其同事在老鼠中找到了类似的种群,称为min,极易患结肠瘤,同时他们还发现另一种群akr,具有抵抗结肠瘤的能力。为进一步弄清其中的缘由,lande等将其进行逆代杂交实验,并分析实验所得数据,检验结肠瘤是否与某遗传基因有关。对每一染色体上的一固定位置x,
引入统计量z(x), 如果在此条染色体上没有变异基因,则在任一位置x, z(x)
服从均值为0的正态分布,但由于假设检验要在整条染色体上进行,发现是否在某一特殊区域内z(x)较大,因而需要知道z(x)沿整条染色体(或其上某一区域)的最大值的分布。lander等证明了在他们所讨论的问题中z(x)
是参数为x=
2的ornstein-uhlenbeck过程。再利用feingold等人(见文献[15])的结果可知:对充分大的t有
其中x(t)是标准正态分布函数,g是染色体的长度。利用上述结果,lander等发现在老鼠的第4条染色体上有一特殊区域与其患结肠瘤有关,假设检验的置信度为0.002[16,
17]。 ※2.1.4 dna序列分析与随机徘徊
dna序列是由a,g,c,t 4个字母组成的序列。1992年voss,
li-kakeko对此做了频谱分析.
同年,peng等的工作揭示了dna序列中存在长程相关而引起人们的兴趣。发现这种相关性的方法是将dna序列表为一维随机徘徊:从第1个碱基(即第1个字母)算起,若是嘌呤碱基(即a或g)则向负走一步,若是嘧啶碱基(即c或t)则向正走一步。记n步后的净位移为fn,
n =1, 2, …, l, l为序列长度.
在长度为l的窗口里计算位移的均方差,然后对全序列求和,得到peng函数f(l)。他们发现,对某种序列(所谓有内含子的序列)有
于是认为这种序列中碱基存在长程相关。长程相关是dna序列分析中的一个研究热点,它可以出现在相隔几千个碱基的位置上。罗辽复把dna序列表为二维随机徘徊,张春霆提出了dna序列的空间曲线表示,都取得了好的成果(见文献[18])。
§2.2 拓扑学方法
dna上碱基的排列次序称为dna的1级结构。双链dna的双螺旋立体结构称为dna的2级结构。双螺旋的中轴线(由每个碱基对的中点所连成的线)也绞拧成螺旋状,称为超螺旋,它可以打结,是dna的3级结构。人类细胞中的46条染色体的dna分子链连起来可达1.8
m,卷曲在细胞核中,就如同200
km长的钓鱼线挤在一个篮球里[19]。研究dna的2级和3级结构,双螺旋及轴线的立体形状、行为以及其生物功能,是非常重要的问题。拓扑学与几何学,特别是纽结理论,是分析此问题的有力武器。
实际上,约在1969年美国拓扑学家fuller,就是应研究dna的分子生物学家的要求而研究闭带形,并得到了与white公式实质上相同的结果。附带谈及,蛋白质也有3级,甚至4级结构。dna中的碱基序列决定蛋白质的1级结构,即氨基酸序列。在合成后,蛋白质便自发折叠成一精确的3级结构,然后才能执行催化、调控、化学输运、流动和结构支持等功能。人们把“dna序列决定氨基酸序列”称为生命的第1密码,而把“蛋白质氨基酸序列决定其自然结构”称为第2密码。破译第2密码的意义十分重大,其中必将用到几何学与拓扑学(参看文献[19])。
§2.3 数理语言学与密码学方法
语言文字是人类表达和传递信息的工具,同样,dna序列也是用以表达和传递人类遗传的信息。
dna这本由30亿个文字(a,g,c,t)写成的无标点、无断句的“天书”是否也应与某种“语言”相对应,如果能掌握它的“语言”就可以读懂它了。美国科学家zipf和shannon用两个标准的语言学实验分析dna,
zipf实验的结果发现“junk”dna与人类的语言具有一样的特征,即单词出现频率的对数与单词排序的对数呈线性关系。shannon的实验的结果也表明“junk”dna有很大的冗余度,这也和人类的语言一致,而dna的编码区则不显示上述任何的语言特征。陈润生等提出用密码学的方法分析dna序列,并取得了一些好结果。语言的数理研究始自chomsky,从形式语言的角度来探讨遗传信息的传递将是很有意义的(详见文献[18]的3.3节彭守礼、刘次全的文章)。
3、基因突变
基因一般是稳定的,但在机体内外因素的影响下,某些基因会发生变异或损伤,基因的突变有时可导致遗传疾病,但也可产生新的种群,从而产生进化。突变与选择是进化的动力。常见的突变是碱基置换(如a,g互换,c,t互换等)和基因缺失,或是各种插入、重复、倒位等。迄今,讨论外源诱导突变的工作较多,如化学诱变、辐射、紫外线照射等。基因的突变是否也有如量子跃迁类似的内秉随机性?
monod认为,dna中的一个突变,以及突变造成的某种蛋白质的过量生产、消失或功能改变属于本质上的偶然性,内在的基因突变,成为密码系统的固有噪音。突变在群体中发生是随机的。
莫诺还认为蛋白质的氨基酸次序也是随机的,他说,“如某种蛋白质含有200个氨基酸残基,即使知道了199个的确切次序,也对剩下的1个不能预测(参见文献[20],p.
71)。突变发生在密码子的第1位,第2位或第3位,概率是不同的,第3位较易突变。从dna到蛋白质,中间要经过rna,因而有不确定性,构成crick摆动。各基因的突变频率不一,例如p53基因是突变最频繁的抑癌基因,它的突变可引起癌症;p73基因与此类似,它位于1号染色体短臂上。tau基因则与痴呆症有关.
关于肿瘤,目前认为它源于某些基因改变,引起细胞突变而异常增殖。单个基因的改变不足以形成肿瘤,肿瘤的发生是多种基因按一定顺序改变的结果。不同肿瘤的点突变方式不同,如肺癌多为g变为t,结肠癌多为g变为a。
关于对基因突变的数学研究,概率统计分析有一些(见文献[1]),但真正有作用的数学模型尚未见到。突变是稀少的,高等生物突变率约在5%—8%。统计物理中研究大概率事件,而生命科学中则多为小概率事件,生命之出现本身就是小概率事件。
除基因突变外,染色体也可以发生畸变。畸变类型主要有两种,一是染色体数目畸变,这时多出或丢失几条染色体;另一种是染色体结构畸变,染色体发生断裂。断裂后的断片未与断端相接而丢失;或断片接同源染色体的相应部分而重复;或断片倒转后接到断端上而使顺序颠倒等等,其结果可能引起先天愚呆、白血病等。
4、结束语
现在,每天得到的生物序列(主要是dna序列、rna序列与蛋白质的氨基酸序列等)的数据量以指数速度增加,按实验室通常处理数据的方法只能处理这些数据中的极小一部分。
2000年6月26日,由美、英、日、法、德和中国组成的国际人类基因组计划协作组分别在六国同时宣布人类基因组工作框架图(覆盖人类基因组90%区域的序列图)绘制完成。如果说,人类基因组是一部蕴涵人类生命奥秘的天书,这一工作意味着人类已经破译了这一天书中的绝大部分文字。这是人类在认识自身,探索生命奥秘的伟大征程中又一里程碑式的工作。
2001年2月15日,人类基因组计划协作组又在世界著名的科学杂志《自然》上联合发表了题为《人类基因组的序列的初步测定及分析》论文,这表明人类已经初步读懂了这部天书的部分内容。整个基因组测序完成后的数据可以构成一本100万页的书,其上只有4个字母的反复出现,既未发现语法,又没标点。如何处理、存储和分析这些数据?
这是数学家、物理学家和生物学家面对的一个难题,需要应用现有的数学方法甚至需要发展新的数学方法与理论来应付这一挑战。另一方面,基因组相关数据库及internet技术的高度发达,使世界各国的科学家都能及时得到待分析的资料与数据,因而从事理论研究的基本条件对所有学者都是相近的,我们应抓住这一大好时机。
不久前,著名的分子生物学家gilbert,
nobel奖获得者在nature上撰文指出,当前分子生物学已进入实验与理论并行发展的阶段。事实上,将概率论与数理统计、计算机等学科应用于分子生物学,经过10多年的发展,一门新兴的学科生物信息学(bioinformatics)已经形成,其研究的主要内容与方法可见文献[21—23].
数学的思想与方法已在物理学中得到广泛应用并获得成功,可以相信在21世纪,其在分子生物学中的应用将会对整个生物学科产生极其深远的影响。
|